
티스토리 뷰
반응형
Suffix tree는 생물학 서열 분석에 있어서 기본적인 데이터 구조 중 하나이다. 이전 블로그에서도 잠깐 언급했듯이 GPU를 이용해서 서열 매칭 작업을 병렬화 한다는 내용을 잠깐 언급한적이 있는데,,,, 이게 좀,,,
Fast Exact String Matching on the GPU와 High-throughput sequence alignment using Graphics Processing Units(MUMmerGPU)는 모두 Michael C. Schatz가 저자로써, 모두 생물학 서열에 있어서 GPU를 이용한(GPU를 사용하지 않는 MUMmer(Maximal Unique Matching)) Suffix tree 알고리즘을 병렬화하는 내용이다. 간단하게 보면, 우선 기준이 되는 Reference Sequence를 로드하고, 이에 대한 Suffix Tree를 생성한다. 그런 후에는 Query Sequence를 로드하고 이를 GPU로 옮긴 후 병렬로(GPU의 각 프로세서에서) 매칭되는 서열을 찾아내고 GPU에서 꺼내와 결과파일을 작성하게 된다.
GPU의 프로세서를 각각의 클러스터 노드로 생각하고 분산되어 서열 매칭 작업을 수행한다고 보면 되겠다. GPU에서는 메모리를 공유하기 때문에 생성된 Suffix Tree를 공유한다는 것이 다르겠지만,,, 코드상에서 심각하게 병렬화라는 단어에 지레 겁먹을 필요없이 작업만 분산된다고 생각하면 되기에 별도의 병렬화에 따른 코드 작성의 어려움은 없으리라 생각된다.
GPU를 이용한 서열 매칭에서의 제한을 잠깐 살펴보면, 논문에서도 성능 비교시 데이터를 보면 각각 Reference Sequence의 길이가 13, 3, 2Mbp로 아주 작은 길이의 서열을 사용하고 있다. 즉, GPU라는 특성상 어쩔 수 없이 Reference Sequence의 길이는 작고(메모리는 적게,,,) 많은 수(2백만개~ 2천6백만개)의 Query Sequence(분산,,,,)를 사용할 수 밖에 없는 영역에서는 뛰어난 능력을 보여 줄 수 있기 때문이다. 실제 서열 검색에 있어서 이러한 (짧은 서열 + 많은 수의 짧은 서열)에 대한것이 얼마나 많은 부분에서 유용할지는 모르겠으나,,, ^^
그럼 좀 더 실용적으로 생각해보면 Reference Sequence의 길이는 Genome-scale을 고려해야 할 것이다. 몇 십Mbp의 수준이 아니라 몇백~몇천Mbp를 염두에 둬야 할 것이다. 인간 염색체 2번을 통째로 올려놓고 몇백만개의 서열에 대해서 매칭 작업을 주욱~할 수 있을 정도로 말이다.
그렇다면 생각해봐야 할것이 어떻게 그렇게 큰 용량을 Suffix tree, 즉 메모리에 올릴 것이냐는 문제인데, 이는 디스크, 가상메모리의 활용 등을 생각할 수 있겠다.
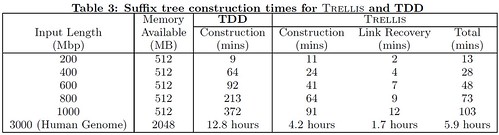
Genome-scale Disk-based Suffix Tree Indexing - Benjarath Phoophakdee를 보면, TRELLIS라는 알고리즘을 통해서 이러한 문제들을 해결하고 있다.
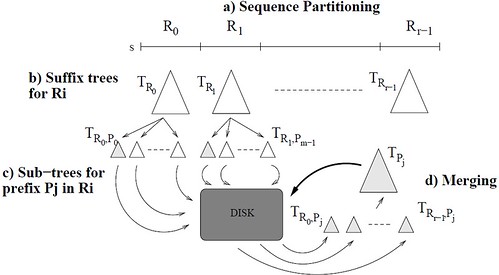
Trellis: 서열을 나누고, 나눈 서열에 대해서 suffix tree를 생성해서 디스크에 쓰고, 합친 후 다시 디스크로, 그럼 큰 길이도 문제없이 suffix tree를 사용
Fast Exact String Matching on the GPU와 High-throughput sequence alignment using Graphics Processing Units(MUMmerGPU)는 모두 Michael C. Schatz가 저자로써, 모두 생물학 서열에 있어서 GPU를 이용한(GPU를 사용하지 않는 MUMmer(Maximal Unique Matching)) Suffix tree 알고리즘을 병렬화하는 내용이다. 간단하게 보면, 우선 기준이 되는 Reference Sequence를 로드하고, 이에 대한 Suffix Tree를 생성한다. 그런 후에는 Query Sequence를 로드하고 이를 GPU로 옮긴 후 병렬로(GPU의 각 프로세서에서) 매칭되는 서열을 찾아내고 GPU에서 꺼내와 결과파일을 작성하게 된다.
GPU의 프로세서를 각각의 클러스터 노드로 생각하고 분산되어 서열 매칭 작업을 수행한다고 보면 되겠다. GPU에서는 메모리를 공유하기 때문에 생성된 Suffix Tree를 공유한다는 것이 다르겠지만,,, 코드상에서 심각하게 병렬화라는 단어에 지레 겁먹을 필요없이 작업만 분산된다고 생각하면 되기에 별도의 병렬화에 따른 코드 작성의 어려움은 없으리라 생각된다.
GPU를 이용한 서열 매칭에서의 제한을 잠깐 살펴보면, 논문에서도 성능 비교시 데이터를 보면 각각 Reference Sequence의 길이가 13, 3, 2Mbp로 아주 작은 길이의 서열을 사용하고 있다. 즉, GPU라는 특성상 어쩔 수 없이 Reference Sequence의 길이는 작고(메모리는 적게,,,) 많은 수(2백만개~ 2천6백만개)의 Query Sequence(분산,,,,)를 사용할 수 밖에 없는 영역에서는 뛰어난 능력을 보여 줄 수 있기 때문이다. 실제 서열 검색에 있어서 이러한 (짧은 서열 + 많은 수의 짧은 서열)에 대한것이 얼마나 많은 부분에서 유용할지는 모르겠으나,,, ^^
그럼 좀 더 실용적으로 생각해보면 Reference Sequence의 길이는 Genome-scale을 고려해야 할 것이다. 몇 십Mbp의 수준이 아니라 몇백~몇천Mbp를 염두에 둬야 할 것이다. 인간 염색체 2번을 통째로 올려놓고 몇백만개의 서열에 대해서 매칭 작업을 주욱~할 수 있을 정도로 말이다.
그렇다면 생각해봐야 할것이 어떻게 그렇게 큰 용량을 Suffix tree, 즉 메모리에 올릴 것이냐는 문제인데, 이는 디스크, 가상메모리의 활용 등을 생각할 수 있겠다.
Genome-scale Disk-based Suffix Tree Indexing - Benjarath Phoophakdee를 보면, TRELLIS라는 알고리즘을 통해서 이러한 문제들을 해결하고 있다.
Trellis: 서열을 나누고, 나눈 서열에 대해서 suffix tree를 생성해서 디스크에 쓰고, 합친 후 다시 디스크로, 그럼 큰 길이도 문제없이 suffix tree를 사용
반응형
공지사항
최근에 올라온 글