
 파이썬을 이용한 개인유전체 분석
파이썬을 이용한 개인유전체 분석
몇년전 개인유전체 데이터가 보편화 되면 자신이 직접 자신의 유전체 데이터를 분석(해킹)할 수 있도록 가이드를 해줄 책을 하나 만들고자 했다. 옛날옛날부터 블로그에 끄적였던 내용들을 정리할겸,, 겸사겸사 이제 책 만들기에는 여러가지 상황상 힘들것 같고, 몇년동안 다른일들 때문에 돌보지 못했던 내용을 하나씩 풀어볼까 한다. 요즘에는 국내에서도 DTC(소비자직접거래) 방식의 유전자 검사를 통해 혈통분석이라는 이름으로 자신의 조상(ancestry)정보 등을 확인할 수 있는데, 오늘은 이와도 연관된 맛보기 챕터로 "인구 집단 비교" 부분을 공개한다. Demo chapter3 from Hong ChangBum 그럼 이만, 뿅
 정밀 종양학 텍스트마이닝(1) - 문헌에서 변이정보 추출하기
정밀 종양학 텍스트마이닝(1) - 문헌에서 변이정보 추출하기
내가 특정 암(또는 질병)에 대한 변이를 모아 놓았다고 할때, 해당 변이가 언급된 논문을 항상 최신으로 업데이트 해주는 경우 해당 변이가 해당 질병에 어떠한 영향을 주는지에 대한 정보를 항상 지켜볼 수 있게 된다. 더 나아가 해당 논문에서 언급된 변이를 논문을 보고 manual curation을 거쳐 knowledge base화 하는데에도 도움을 줄 수 있다. 다음은 BRCA Exchange로 BRCA1/2 mutation의 pathogenicity 정보를 제공하는 데이터베이스로 mutation을 클릭하면 다양한 정보와 함께 해당 mutation이 언급된 논문(제목, 저자, PMID, 해당 mutation이 언급된 본문 내용) 정보를 함께 제공한다. 아래 변이는 BRCA1의 L1750P 변이로 ClinV..
 바이오마커와 정밀종양학(precision oncology)
바이오마커와 정밀종양학(precision oncology)
문헌데이터의 중요성 생물학 문헌 데이터들은 유전자, 단백질(Protein), 화학 성분(Chemical compound) 등 질병 관련 연구에 있어서 중요한 내용을 포함하고 있만 데이터의 양이 방대 하고 산재되어 있어, 연구자들이 일일이 모든 문헌 데이터 를 확인하는 것은 거의 불가능하다. 정밀종양학에 한정하여 환자의 diagnostic, prognostic, predisposing, drug response marker와 gene, variant와의 관계로 한정하여 보면, 1) BRAF V600E와 관련된 논문은 pubmed에서 2004년 5건에서 2017년 454건으로 증가 2) Oncology trial(임상시험)에서 biomarker를 이용한 시험은 전체 시험대비 2000년 ~15%에서 2018년..
들어가는말, 분석 프로토콜과 파이프라인 유전체 데이터 그 중에서도 NGS 데이터 분석에 있어서 많은 툴들이 존재합니다. 이러한 툴을 어떠한 순서로 사용하여 분석하느냐는 바로 분석 프로토콜이 되겠습니다. 흔히 알려진 프로토콜로는 resequecning 데이터를 분석하는 GATK Best Practices가 있습니다. RNA-Seq 데이터 분석에는 Tuxedo protocol이 유명하죠. 연구자들 사이에서는 이거이거이거 사용했더니 좋은 것 같아,,,라는 말들을 자주하곤 합니다. 그러면 연구자는 한번 그 툴들을 사용하여 분석해보죠. 근데 이분석이라는게 한가지 툴로 끝나는게 아니라 툴들을 각각 돌리다 보면 프로토콜의 automation, flexibility, extensionality (자동화, 유연성, 확장..
 진정한 경진대회의 의미를 살린다면...
진정한 경진대회의 의미를 살린다면...
이제 설이다. 새해 복많이 받으세요!!! 몇년전 KOBIC에서 진행한 경진대회가 있었는데,,, 이번에 또 경진대회가 하나 나왔습니다. 아니 나온지 좀 됐습니다. 잠깐 경진대회 이야기나 하고 설맞으로 가려고한다. KOBIC의 생명정보 분석 경진대회 지난 12월 31일까지 접수가 마감이었는데, 1월 15일까지 기간이 연장되었다. 경진대회의 목적은 "NGS 기반 유전체 연구의 활성화 및 발전을 도모하고자 생명정보 데이터 분석 및 알고리즘 개발 경진대회를 진행한다는 것이다" 그리고 그 결과는 논문은로 3월말까지 제출되어야 한다고 한다. 경진대회 홈페이지 https://www.kobic.re.kr/newkobic_competition/ 자 1월15일까지 연구계획서를 제출하고 그걸 3월까지 논문으로 만들어서 제출해..
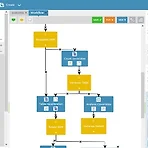 내맘대로 비교/소개하기 - Bioinformatics Big Data
내맘대로 비교/소개하기 - Bioinformatics Big Data
제맘대로 업체선정에서 비교까지 지극히 객관적인 사실이 아닌 주관적으로 비교해 보려고 합니다. 오늘은 그 첫번째 시간으로 Big Data의 관점에서 Bio데이터에 접근하고 그 솔루션을 제공하는 업체 2개를 선택하여 비교하도록 하겠습니다. 우선 선정된 업체 박수로 맞이 하도록 하겠습니다. 두 업체는 국내/국외 업체로 InfiniBio(인피니바이오, 인피니밴드 아닙니다.)와 BioDatomics(바이오데이토믹스)입니다. 둘다 생소한 업체인데요. 아마 다른분들도 다 생소하실거라 생각됩니다. BioDatomics - The Next Generation in Bioinformatics Tools B사는 BioDT라는 툴을 통해 사용자가 NGS 데이터를 핸들링 하도록 하고 있습니다. BioDT는 라이센스 및 사용방법..