
티스토리 뷰
반응형
Gbrowse에서는 GFF 형식의 데이터를 불러 들여 이를 MySQL 데이터베이스에 넣어서 관리한다. 본 글에서는 데이터를 GFF데이터를 읽어 MySQL에 넣는 방법과 MySQL에 저장된 데이터는 어떤 테이블에 어떤 구조로 들어가는지를 살펴보고, 쿼리를 통해서 MySQL에 저장된 데이터를 불러와 이를 Visualization하는 일련의 과정에 대해서 살펴보도록 하겠다.
1. GFF 데이터를 MySQL에 입력하기
BioPerl 패키지에는 GFF 형태의 데이터를 Gbrowse에서 사용할 수 있도록 MySQL에 넣어 주는 bulk_load_gff.pl 파일을 이용하면 된다.
# bulk_load_gff -c -d dbi:mysql:gbrowse_db --user root --password passwd --gff_munge --maxfeautre 1000000000 --local --fasta dna/* *.gff*
위의 명령은 gbrowse_db 데이터베이스에 root 유저와 passwd를 이용하여 접속한 후 dna 디렉토리 밑의 fasta 파일과 현재 디렉토리의 gff를 포함한 GFF 포맷의 파일을 gbrowse에서 사용할 수 있도록 넣으라는 명령이다. 물론 이 명령 전에 gbrowse_db라는 이름의 MySQL 데이터베이스는 생성해야한다.
명령을 수행하면 총 6개의 테이블이 생성되면서 GFF 파일의 내용이 파싱되어 저장된다. 주의할 점은 DB에 넣기전에 tmp 파일들이 생성되는데, 기본적으로 /usr/tmp에 생성된 후 모든 작업이 끝나면 삭제된다.(GFF 파일의 양에 따라 충분한 tmp 용량을 확보할 것)
2. 생성된 테이블 구조
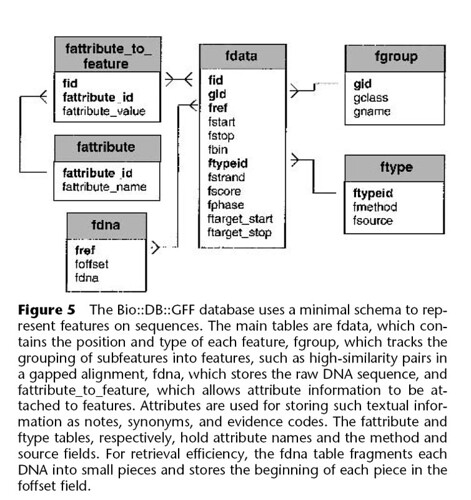
위의 스크립트 후 생성된 테이블과 저장되는 데이터에 대해서 살펴보자. 우선 생성되는 테이블은 위에서 언급했듯이 총 6개로 아래 그림과 같다.
ftype 테이블
ftype 테이블에는 GFF파일의 source와 type 컬럼의 내용이 들어간다. 아래의 경우 type은 snp이고, source는 dbsnp이므로 ftypeid에는 자동으로 할당된 값(1), fmethod에는 snp, fsource에는dbsnp가 들어가게 된다.
GFF형식
seqid source type start end score strand phase attribute <-GFF3 형식
Chr1 dbsnp snp 200 300 . + . ID=SNP:rs1234;alleles:G/T <-GFF3 형식으로 SNP를 표현 1
Chr1 dbsnp snp 400 700 . + . ID=SNP:rs4321;alleles:G/T <-GFF3 형식으로 SNP를 표현 1
fgroup
gclass에는 attribute의 ID다음에 오는 SNP가 gname에는 rs1234가 저장되고 이것에 대한 gid가 자동으로 부여된다.
fattribute
attribute에서 ID를 제외한 부분의 속성들이 저장되며, 위의 예에서는 attrubute가 alleles 하나이므로 fattribute_name에는 allele가 저장되고, 이 속성에 대한 fattribute_id가 자동으로 부여된다.
fattribute_to_feature
실제 속성들의 값이 저장되며, fattribute_id에는 fattribute 테이블에서의 fattribute_id에 대한 실제 값이 fattribute_value에 저장된다. allele라는 속성에 대한 fattribure_id가 6이라면, fattribure_id에는 6, fattribute_value에는 G/T가 저장된다.
fdata
fid는 자동으로 할당된 값, fref는 GFF의 seqid값인 Chr1, fstart는 start값이 200, fstop에는 end값인 300, fbin은 R-Tree에 의해 계산된 bin값과 ftypeid는 ftype테이블, gid는 fgroup의 gid각 각각 저장된다.
상당히 길게 적어놨지만, 아래 E-R을 보면 쉽게 이해가 가실듯,,,
3. 데이터 쿼리
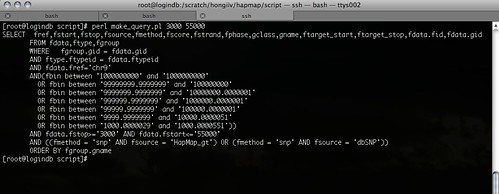
그럼 저렇게 저장된 정보들을 일정영역만 가져오도록 쿼리를 작성하자, 아래 포스팅에서 언급했듯이 R-Tree 알고리즘을 이용해서 저장된 데이터이기 때문에 역시 R-Tree 알고리즘으로 쿼리를 작성해서 가져오면 상당히 빨리 원하는 영역의 데이터를 가져올 수 있다.
R-Tree에서 원하는 영역에 해당하는 bin 쿼리를 만들기 위해서는 Bio/DB/GFF/Adaptor의 dbi.pm의 bin_query() 함수에 찾을 영역의 시작과 끝을 인자로 주면 bin 쿼리가 생성된다. 아래는 영역의 시작과 끝을 입력하면 bin 쿼리와 함께 gbrowse 쿼리를 만들어 주는 스크립트를 실행한 결과이다. 이 쿼리에 대한 결과를 가지고 그래픽을 통해 보여주면 된다.
1. GFF 데이터를 MySQL에 입력하기
BioPerl 패키지에는 GFF 형태의 데이터를 Gbrowse에서 사용할 수 있도록 MySQL에 넣어 주는 bulk_load_gff.pl 파일을 이용하면 된다.
# bulk_load_gff -c -d dbi:mysql:gbrowse_db --user root --password passwd --gff_munge --maxfeautre 1000000000 --local --fasta dna/* *.gff*
위의 명령은 gbrowse_db 데이터베이스에 root 유저와 passwd를 이용하여 접속한 후 dna 디렉토리 밑의 fasta 파일과 현재 디렉토리의 gff를 포함한 GFF 포맷의 파일을 gbrowse에서 사용할 수 있도록 넣으라는 명령이다. 물론 이 명령 전에 gbrowse_db라는 이름의 MySQL 데이터베이스는 생성해야한다.
명령을 수행하면 총 6개의 테이블이 생성되면서 GFF 파일의 내용이 파싱되어 저장된다. 주의할 점은 DB에 넣기전에 tmp 파일들이 생성되는데, 기본적으로 /usr/tmp에 생성된 후 모든 작업이 끝나면 삭제된다.(GFF 파일의 양에 따라 충분한 tmp 용량을 확보할 것)
2. 생성된 테이블 구조
위의 스크립트 후 생성된 테이블과 저장되는 데이터에 대해서 살펴보자. 우선 생성되는 테이블은 위에서 언급했듯이 총 6개로 아래 그림과 같다.
ftype 테이블
ftype 테이블에는 GFF파일의 source와 type 컬럼의 내용이 들어간다. 아래의 경우 type은 snp이고, source는 dbsnp이므로 ftypeid에는 자동으로 할당된 값(1), fmethod에는 snp, fsource에는dbsnp가 들어가게 된다.
GFF형식
seqid source type start end score strand phase attribute <-GFF3 형식
Chr1 dbsnp snp 200 300 . + . ID=SNP:rs1234;alleles:G/T <-GFF3 형식으로 SNP를 표현 1
Chr1 dbsnp snp 400 700 . + . ID=SNP:rs4321;alleles:G/T <-GFF3 형식으로 SNP를 표현 1
fgroup
gclass에는 attribute의 ID다음에 오는 SNP가 gname에는 rs1234가 저장되고 이것에 대한 gid가 자동으로 부여된다.
fattribute
attribute에서 ID를 제외한 부분의 속성들이 저장되며, 위의 예에서는 attrubute가 alleles 하나이므로 fattribute_name에는 allele가 저장되고, 이 속성에 대한 fattribute_id가 자동으로 부여된다.
fattribute_to_feature
실제 속성들의 값이 저장되며, fattribute_id에는 fattribute 테이블에서의 fattribute_id에 대한 실제 값이 fattribute_value에 저장된다. allele라는 속성에 대한 fattribure_id가 6이라면, fattribure_id에는 6, fattribute_value에는 G/T가 저장된다.
fdata
fid는 자동으로 할당된 값, fref는 GFF의 seqid값인 Chr1, fstart는 start값이 200, fstop에는 end값인 300, fbin은 R-Tree에 의해 계산된 bin값과 ftypeid는 ftype테이블, gid는 fgroup의 gid각 각각 저장된다.
상당히 길게 적어놨지만, 아래 E-R을 보면 쉽게 이해가 가실듯,,,
3. 데이터 쿼리
그럼 저렇게 저장된 정보들을 일정영역만 가져오도록 쿼리를 작성하자, 아래 포스팅에서 언급했듯이 R-Tree 알고리즘을 이용해서 저장된 데이터이기 때문에 역시 R-Tree 알고리즘으로 쿼리를 작성해서 가져오면 상당히 빨리 원하는 영역의 데이터를 가져올 수 있다.
R-Tree에서 원하는 영역에 해당하는 bin 쿼리를 만들기 위해서는 Bio/DB/GFF/Adaptor의 dbi.pm의 bin_query() 함수에 찾을 영역의 시작과 끝을 인자로 주면 bin 쿼리가 생성된다. 아래는 영역의 시작과 끝을 입력하면 bin 쿼리와 함께 gbrowse 쿼리를 만들어 주는 스크립트를 실행한 결과이다. 이 쿼리에 대한 결과를 가지고 그래픽을 통해 보여주면 된다.
bin 쿼리와 gbrowse 쿼리 결과
반응형
공지사항
최근에 올라온 글