
티스토리 뷰
반응형
몇 일전에 23andMe가 세번째 버전의 칩을 통해서 좀 더 많은 유전적 정보를 제공한다는 기사가 나왔다. 그럼 본론으로 들어가서 23andMe는 각 고객들간의 유전적 거리를 다음과 같이 2차원의 좌표상에 제공하는데, 기본적으로 칩 컨텐츠에 들어있는 60만개의 SNP 정보를 xy의 두개의 값으로 표현하게 된다.
 |
 |
이러한 방법은 PCA나 MDS와 같은 방법을 이용하는데, 이에 대한 설명은 이전 포스팅을 참고하면 좀 도움이 될듯하다. 간단히 말해서 많은 변수로 이루어진 예를 들어 음악의 경우 곡의재생시간, 가수, 장르, 작곡자, 작사자, 빠르기 등등의 수많은 변수를 간단히 xy의 값으로 축약해서 해당 음악의 특성을 한눈에 볼 수 있도록 하는 것이다. 그렇다면 SNP 데이터의 경우 어떤 데이터를 어떻게 가공해서 이를 축약하는지 그리고 이 축약된 데이터를 어디에 활용하는지에 대해서 알아보겠다.
기초 Genome 이론: IBD vs. IBS
IBD (Identity by Descent)는 두 사람이상이 조상(부모)으로 부터 어떠한 allele를 물려 받았는지를 통해 서로간의 위치를 학인하는 방법이며, IBS (Identity by State)는 두 사람간에 어떠한 allele를 공유하는지를 단순히 말해주게 된다.
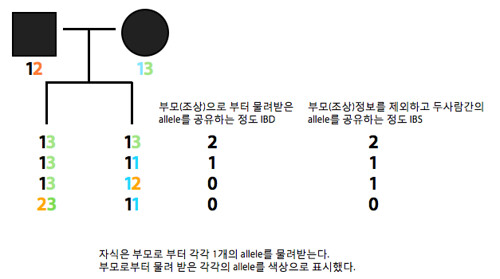
첫번째 라인의 자식들은 각각 아버지로부터 1을 어머니로부터 3을 물려받았다. 따라서 13/13으로 각각 부모로 물려받은 공통 allele의 갯수가 2개이므로 IBD 값은 2, 부모를 배제하고 서로간의 13/13 두개의 allele가 같으므로 IBS 값도 2가 된다. 세번째 라인의 경우 13/12 로 서로 같은 1을 가지고 있지만 이는 각각 부/모로 부터 따로 받은 것이기 때문에 부모로부터 받은 allele를 서로간에 공유하는것이 없기 때문에 IBD는 0이지만, 서로간에 1이라는 것을 공유하기 때문에 IBS는 1이 된다.
이러한 IBD나 IBS 값을 모든 사람들간에 pairwise로 비교하면 모든 사람들간의 유전적인 거리를 알 수 있게 되는데 이러한 IBD/IBS 값을 가지고 PCA나 MDS를 수행하게 된다. 그러나 이러한 비교는 비교할 SNP(비교할 allele)의 갯수가 백만개에 비교할 사람이 만명이 된다면 이를 계산하는데에는 엄청난 컴퓨팅 시간이 소요된다. 일례로 KARE 프로젝트의 샘플(50만개 SNP에 대해서 8842명을 pairwise로 비교)의 경우 IBS 값을 계산하는데 컴퓨터 몇 일이 소요되었다.
Parallelize IBD estimation
이러한 IBD를 계산하는데에는 PLINK라는 소프트웨어서 가능하며, 각각의 비교를 클러스터컴퓨터와 스케줄러(PBS, Torque, SUN Grid Engine 등)를 통해서 수행하게 되면 linear한 시간에 계산을 수행할 수 있게 된다. 우선 PLINK 포맷의 SNP 데이터를 준비한다. PLINK에서 사용하는 Binary 포맷은 bed, bim, fam의 3개의 확장자를 가진 포맷으로 되어 있으며, bed 파일은 Binary 형태의 SNP genotype 데이터, bim 파일은 SNP의 annotation 정보, fam 파일은 샘플의 정보가 들어있다.
PLINK에서는 --genome-list라는 옵션을 통해서 샘플을 잘라서(split) 사용할 수 있도록 해주는데, UNIX의 split 명령을 통해서 fam 파일의 Family ID와 Individual ID만을 1000명씩 포함하도록 한다. split 명령을 통해서 8842명의 데이터를 1000명씩 잘라서 넣은 총 8개의 tmp.list번호 형태의 파일(small chunk)이 생성된다.
이렇게 생성된 파일은 PLINK의 --genome 옵션을 통해서 IBD 값을 계산하게 되면 이때 전체 frequency 데이터를 --read-freq 옵션을 통해 지정해 주고, 만들어진 chunk 들을 pairwise로 모두 계산되도록 아래와 같이 PLINK 명령어를 만든다. 전체 frequence(KARE_8842.frq)는 다음과 같은 PLINK 명령을 통해서 얻는다.
plink --bfile KARE_8842 --freq --out KARE_8842

이렇게 만들어진 plink 명령어를 PBS나 Torque와 같은 스케줄러를 통해 알맞게 클러스터의 노드들에서 계산하도록 한 후 만들어진 결과 파일들을 하나로 묶어주면 모든 individual에 대해서 pairwise로 비교한 IBD 값을 얻을 수 있다.
만들어진 output 파일은 다음과 같다.
data.sub.1.1.genome
data.sub.1.2.genome
data.sub.1.3.genome
data.sub.1.4.genome
...............
data.sub.8.8.genome
생성된 output 파일을 하나의 파일로 만들고 중간에 생성된 파일들은 삭제하면 된다.
cat data.sub.*genome > results.genome
rm tmp.list*
rm data.sub.*
이상의 fam파일을 split하고 스케줄러 스크립트와 output 파일을 merge하는 스크립트는 Getting Genetics Done의 "Parallelize IBD estimation with PLINK" 포스팅 참고하면 된다.
다음 (2)편에서는 IBD, IBS를 실제 클러스터상에서 실행하고 이를 활용하는 방법에 대해서 살표보겠다. -.-;;
기초 Genome 이론: IBD vs. IBS
IBD (Identity by Descent)는 두 사람이상이 조상(부모)으로 부터 어떠한 allele를 물려 받았는지를 통해 서로간의 위치를 학인하는 방법이며, IBS (Identity by State)는 두 사람간에 어떠한 allele를 공유하는지를 단순히 말해주게 된다.
첫번째 라인의 자식들은 각각 아버지로부터 1을 어머니로부터 3을 물려받았다. 따라서 13/13으로 각각 부모로 물려받은 공통 allele의 갯수가 2개이므로 IBD 값은 2, 부모를 배제하고 서로간의 13/13 두개의 allele가 같으므로 IBS 값도 2가 된다. 세번째 라인의 경우 13/12 로 서로 같은 1을 가지고 있지만 이는 각각 부/모로 부터 따로 받은 것이기 때문에 부모로부터 받은 allele를 서로간에 공유하는것이 없기 때문에 IBD는 0이지만, 서로간에 1이라는 것을 공유하기 때문에 IBS는 1이 된다.
이러한 IBD나 IBS 값을 모든 사람들간에 pairwise로 비교하면 모든 사람들간의 유전적인 거리를 알 수 있게 되는데 이러한 IBD/IBS 값을 가지고 PCA나 MDS를 수행하게 된다. 그러나 이러한 비교는 비교할 SNP(비교할 allele)의 갯수가 백만개에 비교할 사람이 만명이 된다면 이를 계산하는데에는 엄청난 컴퓨팅 시간이 소요된다. 일례로 KARE 프로젝트의 샘플(50만개 SNP에 대해서 8842명을 pairwise로 비교)의 경우 IBS 값을 계산하는데 컴퓨터 몇 일이 소요되었다.
Parallelize IBD estimation
이러한 IBD를 계산하는데에는 PLINK라는 소프트웨어서 가능하며, 각각의 비교를 클러스터컴퓨터와 스케줄러(PBS, Torque, SUN Grid Engine 등)를 통해서 수행하게 되면 linear한 시간에 계산을 수행할 수 있게 된다. 우선 PLINK 포맷의 SNP 데이터를 준비한다. PLINK에서 사용하는 Binary 포맷은 bed, bim, fam의 3개의 확장자를 가진 포맷으로 되어 있으며, bed 파일은 Binary 형태의 SNP genotype 데이터, bim 파일은 SNP의 annotation 정보, fam 파일은 샘플의 정보가 들어있다.
PLINK에서는 --genome-list라는 옵션을 통해서 샘플을 잘라서(split) 사용할 수 있도록 해주는데, UNIX의 split 명령을 통해서 fam 파일의 Family ID와 Individual ID만을 1000명씩 포함하도록 한다. split 명령을 통해서 8842명의 데이터를 1000명씩 잘라서 넣은 총 8개의 tmp.list번호 형태의 파일(small chunk)이 생성된다.
gawk '{print $1,$2}' KARE_8842.fam | split -d -a 3 -l 100 - tmp.list이렇게 생성된 파일은 PLINK의 --genome 옵션을 통해서 IBD 값을 계산하게 되면 이때 전체 frequency 데이터를 --read-freq 옵션을 통해 지정해 주고, 만들어진 chunk 들을 pairwise로 모두 계산되도록 아래와 같이 PLINK 명령어를 만든다. 전체 frequence(KARE_8842.frq)는 다음과 같은 PLINK 명령을 통해서 얻는다.
plink --bfile KARE_8842 --freq --out KARE_8842
이렇게 만들어진 plink 명령어를 PBS나 Torque와 같은 스케줄러를 통해 알맞게 클러스터의 노드들에서 계산하도록 한 후 만들어진 결과 파일들을 하나로 묶어주면 모든 individual에 대해서 pairwise로 비교한 IBD 값을 얻을 수 있다.
만들어진 output 파일은 다음과 같다.
data.sub.1.1.genome
data.sub.1.2.genome
data.sub.1.3.genome
data.sub.1.4.genome
...............
data.sub.8.8.genome
생성된 output 파일을 하나의 파일로 만들고 중간에 생성된 파일들은 삭제하면 된다.
cat data.sub.*genome > results.genome
rm tmp.list*
rm data.sub.*
이상의 fam파일을 split하고 스케줄러 스크립트와 output 파일을 merge하는 스크립트는 Getting Genetics Done의 "Parallelize IBD estimation with PLINK" 포스팅 참고하면 된다.
다음 (2)편에서는 IBD, IBS를 실제 클러스터상에서 실행하고 이를 활용하는 방법에 대해서 살표보겠다. -.-;;
반응형
공지사항
최근에 올라온 글