
 바이오마커와 정밀종양학(precision oncology)
바이오마커와 정밀종양학(precision oncology)
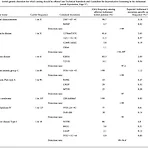
문헌데이터의 중요성 생물학 문헌 데이터들은 유전자, 단백질(Protein), 화학 성분(Chemical compound) 등 질병 관련 연구에 있어서 중요한 내용을 포함하고 있만 데이터의 양이 방대 하고 산재되어 있어, 연구자들이 일일이 모든 문헌 데이터 를 확인하는 것은 거의 불가능하다. 정밀종양학에 한정하여 환자의 diagnostic, prognostic, predisposing, drug response marker와 gene, variant와의 관계로 한정하여 보면, 1) BRAF V600E와 관련된 논문은 pubmed에서 2004년 5건에서 2017년 454건으로 증가 2) Oncology trial(임상시험)에서 biomarker를 이용한 시험은 전체 시험대비 2000년 ~15%에서 2018년..
 Deep Genomics
Deep Genomics
누가 뭐래도 요즘 화두는 deep learning이 아닐까? 그렇다면 현재 genomics 또는 이와 관련하여 어떤 움직임들이 있는지 한번 알아보도록 한다. 유전질환 딥러닝 - Face2Gene 예전 칼 짐머의 "Game of Genomes"의 시즌1의 두번째 에피소드인 "깨어진 코드"편을 보면 이런 장면이 나온다. 칼 짐머가 자신의 염기서열을 시퀀싱 하기로 한후 보스턴의 브리검여성병원의 로버트 그린은 칼 짐머의 얼굴을 유심히 보는 장면이 나온다. “전 지금 유전병에서 나타나는 얼굴의 특징을 찾고 있는 거에요” 브리검여성병원(Brgham and women’s hospital)의 로버트 그린이 말했다. “눈의 모양, 귀가 너무 낮게 있지는 않은지. 귀가 복잡하게 생기진 않았는지” 그린박사는 사무실을 앞뒤로..
 23andMe 재단장
23andMe 재단장
국내 질병 예측성 유전자 검사 현황 지난 19일 "질병예측성 유전자 검사의 개선방안 공청회"가 진행되었다. 공청회의 질병관리본부의 질병예측성 유전자 검사의 관리에 관한 용역 결과에 대한 세션에서 의하면 유전자 검사를 6개의 카테고리로 나누어 관리하는 방안으로 카테고리 1~4까지는 기존의 유전자 검사에 해당하며 카테고리 5와 6의 경우는 다음과 같은 기준으로 나뉘어 관리하도록 하고 있다. 질병관리본부의 질병예측성 유전자 검사 관리 용역 결과 유전자 검사 분류 이에 유전체기업협의회의 경우 기존 검사와 더불어 질병예측성 유전자검사와 웰니스 유전자 검사의 카테고리로 나누어 관리하고자 하고 있다. 특히 웰니스 유전자 검사의 경우 DTC를 허용하는 것을 골자로 하고 있다. 유전체기업협의회의 유전자검사 분류 방안 미..
 결함을 허용하는 의존성 있는 태스크의 관리
결함을 허용하는 의존성 있는 태스크의 관리
그래프를 이용한 태스크 표현 흔히 바이오인포매틱스 분석이라고 하는 경우 스크립트나 모듈을 작성하여 일련의 분석을 수행하곤 한다. 그러나 단순한 형태의 일이 아니라 더욱 고난이도의 일을 처리하다가 보면 (물론 대부분이 그렇지만) 태스크의 의존성을 고려해야 하는 경우가 많다. 그래프를 이용하여 이를 표현해 보면서 어떻게 의존성과 결함을 고려한 스케줄러를 만들 수 있는지 생각해 보도록 하자. 총 4개의 노드 (Task1, Task2, Task3, Task4)와 엣지로 구성된 그래프로 각각의 엣지는 다음과 같은 의존성을 가진다. Task1=>[Task2, Task3] Task2=>[Task4] Task3=>[Task4] Task4=>[] 즉, Task1이 끝나야 Task2,3이 수행되고 Task4는 Task2,..
 BGZF (Blocked GNU Zip Format)
BGZF (Blocked GNU Zip Format)
Random Access BAM 파일의 경우에는 BGZF를 이용하기 때문에 원하는 곳으로 빠르게 access가 가능하다. 우리가 흔히 사용하는 GZIP (GNU ZIP) 보다는 압축효율 (압축했을때 용량)이 떨어지지만 random access가 가능하다는 잇점으로 인해 BAM 파일(BAM의 경우 재빠르게 자신이 원하는 position을 뷰잉하는데 많이 사용하기 때문)에서 사용하는 기술이다. 용량이 큰 텍스트 파일을 압축해 놓고 파일의 어느 부분이던지 랜덤하게 액세스 가능하기 때문에 그 활용도가 높은데 특히나 클러스터를 이용하는 경우 파일을 분할하는 등의 I/O 작업이 필요 없기 때문에 그 활용이 매우 높다고 할 수 있다. 여러 활용중 하나로 FASTQ 파일에 적용하여 사용하고 있다. FASTQ 응용 일루..
 Split Reads
Split Reads
Split Read (SR) Split Read(SR)는 하나의 read가 분리 (split)된 것으로, 여기서 분리는 read의 일부분이 reference에 align되고 나머지 일부분은 또 reference의 다른 부분에 align된 것으로 Chimeric Alignment라고도 한다. SR은 deletion, insertion, inversion, tandem duplication과 같은 structural variation을 찾는데 유용한 지표로 사용된다. Identification of a deletion in an individual genome by split read analysis SAM에서의 SR 흔적 SAM파일에서는 SR을 표시하는데 SA 태그를 사용한다. SA 태그는 Chimeric..